E&ES 359: Climate Change
Mass spectrometry
The element oxygen occurs as 3 stable isotopes: the common isotope 16O (99.765%), the rare isotope 18O (0.1995%), and the very rare isotope 17O (0.0355%). The element carbon occurs in two stable isotopes (in addition to the radioactive isotope 14C): the common isotope 12C (98.89%), and the rare isotope 13C (1.11%). Usually, the common isotope is lighter than the rare ones. An element must be in the gas phase if we want to measure its isotopic ratios in a mass-spectrometer, and the gas used for analysis of oxygen isotopes is CO2.
A CO2 molecule can thus have different molecular weights (the parameter measured in a mass-spectrometer) as a result of the presence of the three stable isotopes of oxygen and two of carbon. The most common configurations of CO2 are: 12C16O16O (molecular weight 44), by far the most common molecule; 13C16O16O (molecular weight 45), and 12C18O16O (molecular weight 46). These are all molecules which have no or only one of the rare isotopes. Molecules in which two rare isotopes are present are extremely rare, because the probability of pairing (for instance) two 18O-atoms in one CO2 molecule by randomly moving atoms is infinitesimally small).
To measure the molecular weight of CO2 in a mass spectrometer, the gas is liberated from CaCO3 by reaction with 100% orthophosphoric acid (H3PO4) at 25oC:
3CaCO3 + 2H3PO4 -> 3CO2 + 3H2O + Ca3(PO4)2.
It is very important to adhere to strictly normalized reaction conditions, because only 2 of the 3 O-atoms in CaCO3 are present in the CO2-gas (the third one moves into a water molecule, with the hydrogen from the acid). If conditions are not normalized, isotopic fractionation will vary from sample to sample. The oxygen isotopic composition of water is measured by equilibration of the water with CO2-gas at 25oC. This makes it possible to compare the oxygen isotopic ratios in a water sample (as obtained from comparison with a water-standard) with isotopic values obtained from calcite (as compared with a calcite standard - see below). Isotopic ratios of carbon and oxygen are thus measured at the same time, on exactly the same material.
Isotope fractionation is the partial separation of isotopes of the same element during physical (e.g., evaporation) or chemical (e.g., precipitation reaction) processes. The separation results from the small differences in physico-chemical properties of molecules containing different isotopes. Kinetic isotopic fractionation occurs when the rates of processes differ between isotopic species (as a result of molecules with the lighter isotope moving faster). Equilibrium isotope fractionation occurs as a result of differences in thermodynamic properties of molecules containing different isotopes. The following reactions are involved in the precipitation of calcite:
Ca2+ + 2HCO3- -> CaCO3 + H2O + CO2,H2O + CO2 -> H+ + HCO3-.
The oxygen in bicarbonate (HCO3- ), the most common carbon-bearing ion in sea water (at its present pH of 8.1-8.2 in surface waters, 7.6 or so in deep water) equilibrates isotopically with the oxygen in water, and the bicarbonate is used by organisms building carbonate shells. Isotope fractionation between two substances (such as the bicarbonate ion dissolved in water and carbonate) accompanying a specific process (such as the precipitation of calcite) is expressed in the fractionation factor (a):
a (c-w) = Rc/Rw,
in which a(c-w) is the fractionation factor , and Rc and Rw are the isotopic ratios (18O/16O) in calcite and dissolved bicarbonate in water, respectively. Note that the abundance of the rare isotope is divided by the abundance of the common isotope, so that R<< 1. There is an inverse relation between the fractionation factor a and temperature, so that fractionation is less at higher temperatures, and a becomes equal to 1 (no fractionation) at high temperatures. According to the equation derived from a study of experimental inorganic precipitation of calcite over a temperature range between 0 and 500oC:
103ln a = 2.78 (T-2.106) -3.39,
where T is the temperature in degrees Kelvin.
Conventions in describing oxygen and carbon isotope data
The absolute abundance of an isotope is difficult to measure with sufficient accuracy, and therefore we compare isotopic ratios in a sample with those in a standard, resulting in the delta-notation:
d(x) = [{Rx - Rst}/Rst] . 103,
where d(x) is the delta-value of a sample, and Rx and Rst are the isotopic ratios in sample and standard, respectively. The d-value is the relative difference in the isotopic ratio of the sample and the standard, expressed in part per mille (o/oo). Carbon and oxygen data from carbonates are usually referred to the PDB standard (a belemnite, Belemnitella americana, from the Late Cretaceous PeeDee Formation in South Carolina), but oxygen isotopic compositions of water samples are referred to standard mean ocean water (SMOW). The scales are related to each other as follows:
d18Ocalcite(vs. SMOW) = 1.03086 . d18Ocalcite (vs. PDB) + 30.86.
Because of the inverse relation between fractionation factor a and temperature, we can use the isotopic composition of a carbonate, precipitated in isotopic equilibrium from water with a known isotopic composition, to deduce the temperature at which the calcite was precipitated. The empirically determined temperature equation that is used most often in work with deep-sea carbonates is:
toC = 16.9 - 4.38 (dc - dw) + 0.10 (dc - dw)2,
in which dc is the oxygen isotopic composition of calcite compared with the PDB standard, dw the oxygen isotopic composition of the water from which it was precipitated compared with the SMOW standard. We measure dc in order to derive toC. At constant dw, a temperature change of about 4oC corresponds to a change in dc of about 1 o/oo; we can measure dc at an accuracy of +0.1 o/oo .
NOTE that we can use this method only IF
Neither of these has to be true for samples from the geological record.
The temperature equation was derived for inorganically precipitated calcite, and most naturally occurring sample material has been precipitated by organisms. For much paleoceanographic work various species of planktonic (floating in the water column) and benthic (living on the sea floor) foraminifera are used. The combination of data on planktonic and benthic species allows determination of temperatures in surface and deep waters.
The largest known, and fastest, effect on isotopic composition of the oceans in the Cenozoic (the last 65 milion years) has been by the volume-changes in the polar ice sheets. Most water on Earth (97.25%) is stored in the oceans, 2.05% in ice (90% of which resides in the Antarctic ice sheet), with the remainder stored as ground water (0.68%), while the amount of water in lakes and rivers, as soil moisture, in the biosphere and atmosphere is insignificant.
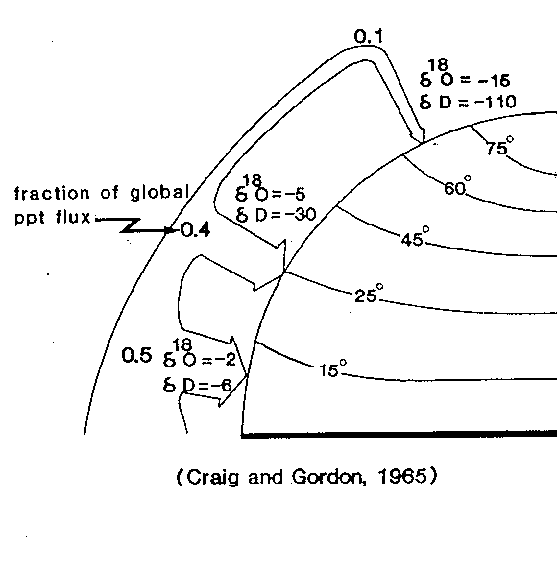
Oxygen isotopic fractionation occurs during evaporation and condensation of water, with enrichment of the heavier isotope in the liquid phase. Water vapor is thus isotopically light compared with the ocean water from which it evaporated, and condensed rain is isotopically heavy compared with the water vapor from which it condensed. Rain that falls inland is therefore more depleted in 18O than rain in coastal areas, because much rain condenses in coastal areas (especially when humid air has to move up to cross mountains), and the remaining water vapor traveling inland has been isotopically depleted.
With much simplification, we can say that in the subtropics evaporation strongly exceeds precipitation, and at temperate to high latitudes the reverse is true (more precipitation than evaporation). Thus there is a net transport of water vapor from subtropical to high latitudes. Water vapor generated in the subtropics is isotopically lighter than the sea-water from which it formed; rain falling out is heavier than the remaining vapor , which thus is depleted even more in the heavy isotope. We can see the Earth as a multiple-stage distillation column, with reflux of the condensate to the reservoir; such a prcoess is called Rayleigh-fractionation. The reservoir is the ocean, rain at different latitudes is the condensate at different stages, and the polar ice (which forms from the snow, formed from vapor that has travelled furthest away from the area of original evaporation) is the highest stage of the column (and is thus the most depleted in the heavy isotope) .
Figure 1: Note that dD stands for the isotope ratio of hydrogen, where the heavy isotope (2H) is called Deuterium; ppt flux means 'precipitation flux).
The snow in the interior of Antarctica (a continent at high latitudes) is thus isotopically the most depleted precipitation on earth, with values as low as -60 o/oo ; in Antarctic coastal areas values are approximately between -17 and -30 o/oo. The average isotopic composition of the Antarctic ice sheet with a volume of about 26 x 106 km3 is not known precisely (the ice sheet is up to 4 km thick), and must be estimated. Probably there is a larger contribution from the more depleted snow inland (the snow falling in the coastal areas changes into ice that is quickly calved off as bergs); estimates are on the order of -35 to -50 o/oo . The present ice sheets are strongly depleted in 18O as compared to ocean water, which has an an average d18O of -0.28 o/oo . As an extimate, the oxygen isotopic composition of the ocean as a whole would be about -1.26 o/oo if all the ice sheets melted. In order to use oxygen isotopic data from carbonates to calculate paleo-temperatures, we thus have to make estimates of the volume of polar ice (e.g., using data on sea-level), and of its isotopic composition.
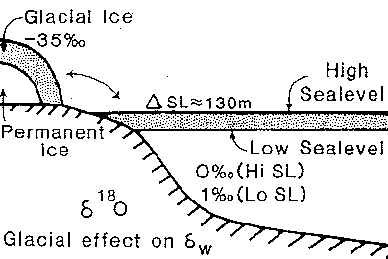
The major problem, as explained above, is that we always see the sum of ice-volume (reservoir) and temperature changes (plus local effects), and have in the past not had many ways to decide which is which. We can make the assumption that co-variance between benthic and planktonic records from low latitudes indicates changes in ice-volume, because ice-volume changes affect the whole ocean, but temperature changes appear more probable in surface than in deep-waters. Recently, efforts have started to estimate paleo-temperatures from other information (the amount of Mg and Sr built into foraminiferal tests); the combination of the paleo-temperature and the oxygen isotope data can then be used to estimate global ice volume.
The Cenozoic deep-sea oxygen isotope record shows the following features. Remember that the deep waters of the ocean form by sinking at high latitudes; the values for benthic foraminifera (in the deep sea) thus reflect the deep-water temperatures globally, as well as the surface water temperatures at high latitudes.
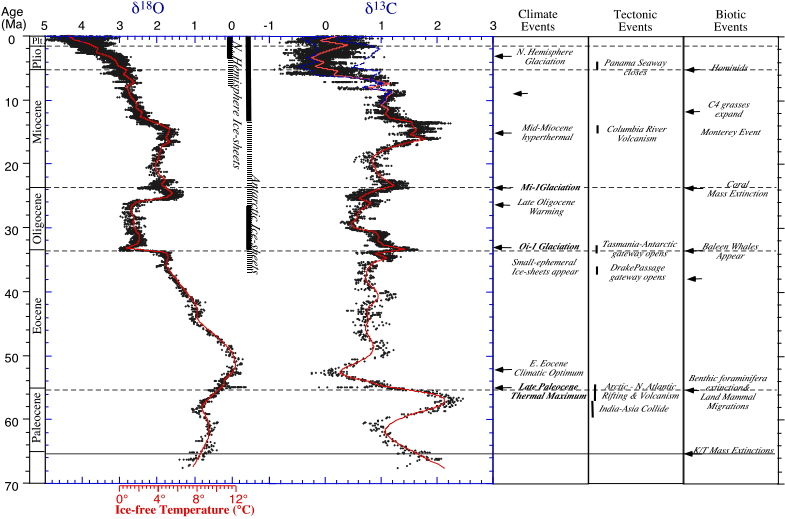
During the Cenozoic (after the Cretaceous/Tertiary boundary at 65 Ma) the d18O values of benthic foraminifera increased, but the values in surface-water planktonic foraminifera at low latitudes (not shown) showed little to no change. The increases did not occur gradually, but in "steps", intervals of sudden change, alternated with stable intervals. Major steps each lasted about 105 years or less, and occurred:
Figure 3: Oxygen isotope values (left column) and carbon isotopic values of the deep sea for the Cenozoic, after Zachos, J., Pagani, M., Sloan, L., Thomas, E., and Billups, K., 2001, Trends, Rhythms, and Aberrations in Global Climate Change 65 Ma to Present. Science, 292: 686-693. Note that lower (more negative) d18O values mean that water temperature was higher, or the polar ice sheets were smaller, or both at the same time. Note temperature scale (given for ice-free conditions) below; see description of events to the right. NOTE: this figure replaces figure 7-7 (p. 153) in your text book; the latter is outdated.
Carbon isotopic records from carbonates are of interest to paleoceanographers because they help understand the functioning of the carbon cycle during earth history. In the carbon isotopic values in carbonates there is not much of a temperature-effect, but there are complex local effects in additition to the global effect of varying amounts of carbon being stored in an isotopically-light reservoir (biosphere).
The two major sedimentary reservoirs of carbon are organic material and calcium carbonate (inorganic carbonate), both are secreted by the biosphere. The precipitation of carbon in carbonate (organically or inorganically) involves only very little isotopic fractionation relative to total dissolved inorganic carbon. The d13C of calcite is relatively insensitive to changes in temperature (about 0.035 o/oo per oC). Therefore the d13C of inorganically precipitated carbonate in the oceans is very close to that of total dissolved carbonate.
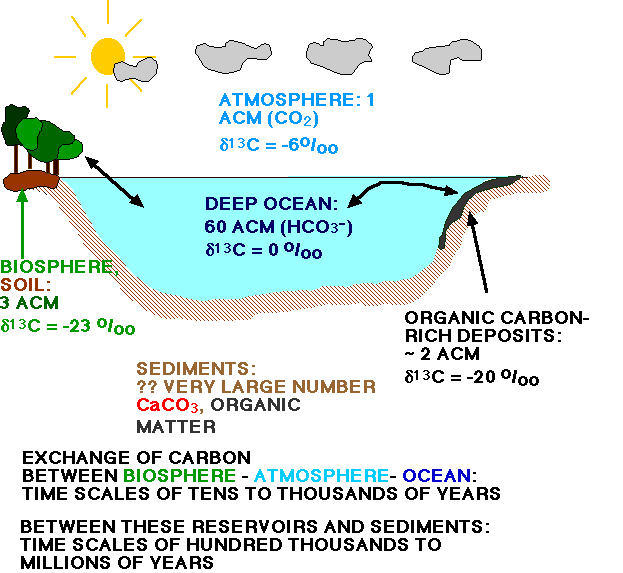
Figure 4 shows the reservoirs where carbon is stored, with notes on time scales at which carbon moves into and out of these reservoirs. You have discussed the long-term carbon cycle in class; the short-term carbon cycle you weill discuss in much more detail. ACM means: Atmospheric Carbon Mass, how much carbon there was in the atmosphere at pre-industrial levels.
THE main fractionating process in the carbon cycle is photosynthesis, because the photo-synthetic fixation of carbon involves a very large fractionation, with organic matter being depleted in the heavy isotope (isotopically light). The simplified reaction for photosynthesis is:
6CO2 + 6H2O -> C6H12O6 + 6O2.
Almost all carbon in organic compounds in the biosphere is ultimately derived from photosynthetically (and a little from chemosynthetically) produced material, thus all organic carbon compounds in the biosphere are isotopically very light.
Average d13C values of organic carbon in land plants vary according to the chemical pathway of photosynthesis: plants using the C3 pathway of photosynthesis (most higher plants) show maximum fractionation, and have isotopic ratios between -23 and -33 o/oo , with an average of about -26 o/oo, while the isotopic composition of CO2 in the atmosphere is about -6 to -7 o/oo. Plants using the C4 pathway (most tropical and marsh grasses; since the Miocene) average about -13 o/oo, ranging from -9 to -16 o/oo . The photosynthetic reaction pathways of marine phytoplankton, which use dissolved CO2 (gas) are not well known, but the organic matter in marine phytoplankton shows a range of values between -10 and -31 o/oo , with most values between -17 and -22 o/oo . These topics will be discussed in more detail later in the class.
There have been large changes over geological time in how much carbon present in the atmosphere-ocean system was put into organic carbon, how much into carbonate (see second curve in figure 3, above). Therefore the amount of 13C available for organisms in the shape of total dissolved inorganic carbon in the oceans (TDIC, mainly HCO3- as explained above) has varied over time (similar to the oxygen isotope concentration varying depending upon the size of the ice caps). Presently over 90% of the carbon in the ocean-atmosphere system resides in the deep ocean (the whole oceanic reservoir is about 60 x as large as the atmospheric reservoir in pre-industrial times), thus we can use the d13C signal from deep waters in the largest ocean, the Pacific, as a proxy for the average ocean d13C signature (Figure 4). The largest sedimentary reservoirs of carbon (output from the ocean) are organic carbon and carbonate carbon. Presently the outflux of carbon from the ocean in calcium carbonate is about 4 x as large as the outflux in organic carbon, and the oceans are about in steady state (or: they would be if there was no fossil fuel burning).
If there are changes in the input or output ratio of organic matter and carbonate carbon, the d13C of TDIC in the whole ocean must change: if more carbon is removed from the oceans in organic matter, then more 12C is removed from the oceans, thus the d13C value of TDIC in the whole ocean increases. Similarly, if there is a period of strong erosion of organic material (e.g., coal) from land and its influx in the oceans, then the average d13C value of the TDIC in the oceans decreases.
Changes in the sizes of these reservoirs occur slowly (on a geologic time-scale, 105 - 106 year), because oceanic deposition and erosion can not be re-organized quickly. For instance, a change in erosion-rate may result from a change in continental elevation, i.e., mountain building processes.
A reservoir that can react quickly (on the order of 102-103 years), however, is the biosphere. Presently, about 80% of all organic carbon is stored on land (and about 78% of all this material is present in soils), about 20% in the oceans (almost 90% of the organic carbon in the oceans is in the deep sea). We expect changes in the terrestrial biosphere mass to be reflected in the carbon isotopic composition of TDIC in the oceans via the d13C of CO2 in the atmosphere, because the "atmosphere is the slave of the ocean" as a result of its much smaller size (the oceanic reservoir is about 60 x as large as the atmospheric reservoir).
Complexity 1: productivity in the oceans
The photosynthetic activity in the oceans (limited to the photic zone) results in a very strong depletion of the surface waters in 12C (captured in organic matter), strong enrichment in 13C. Planktonic organisms living in the photic zone, and forming calcareous tests from dissolved inorganic carbon (largely HCO3- ) in these surface layers, thus use carbon that is enriched in 13C (isotopically heavy) for the formation of their tests. Common values recorded in the present oceans for planktonic foraminiferal tests are about +2 to +3 o/oo , while the average whole ocean d13C value of the total dissolved inorganic carbon is around 0.
Figure 5. Effects of the biological pump on carbon isotopic composition.
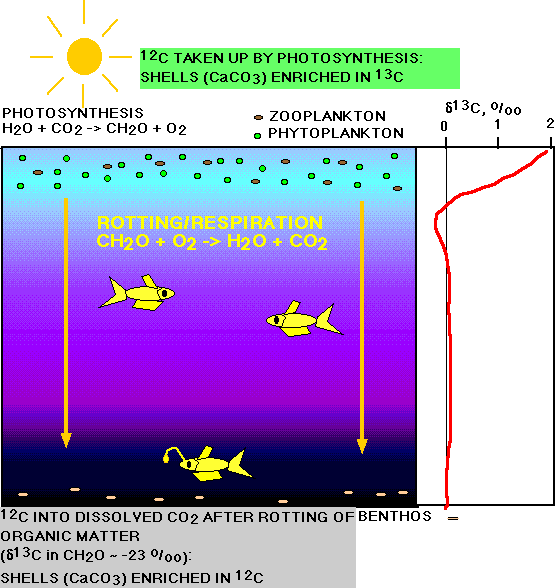
The d13C values of dissolved inorganic carbon at greater depths (and thus the d13C values of benthic foraminifera) are quite different from those in surface waters, because carbon dioxide derived from oxidized organic material (thus isotopically light) is added to the deep waters.
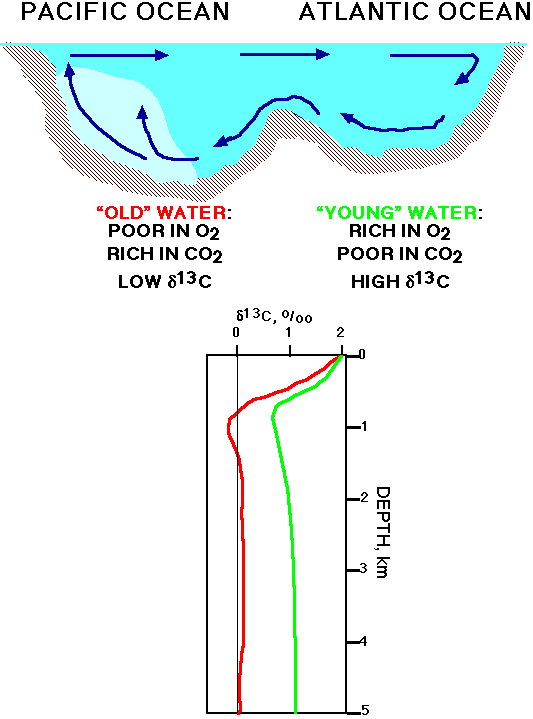
Complexity 2: ocean circulation
To complicate factors more, there is a difference in d13C of total dissolved inorganic carbon (TDIC) in deep waters with a different "age": waters that have been out of contact with the surface ocean for a long time, have accumulated much carbon derived from oxidation of organic material, thus their total dissolved inorganic carbon is isotopically light. In the present oceans there is a difference in isotopic composition of TDIC in the deep Atlantic (younger waters) and the deep Pacific (older waters; fig. 5), with the north Pacific being about 1 o/oo lighter than the Atlantic (Figure 5).
The activity of the biological pump, photosynthesis in surface water, dead organic matter sinking to the bottom, thus causes the existence of a gradient in d13C values between surface and deep waters, with the TDIC in surface waters isotopically heavier than in deep waters.
The actual vertical gradient at a specific location depends on a several factors, including the primary productivity of the surface waters (higher productivity -> more 12C stored in organic matter -> isotopically heavier TDIC -> heavier CaCO3 tests of planktonic foraminifers). It also depends on the age of the deep water at the site, which depend upon the location and the circulation pattern of the oceans.
Figure 6: Carbon isotopic composition of total dissolved inorganic carbon in Atlantic and Pacific Oceans.
The oceanic record of d13C in planktonic and benthic foraminifera at any one site always reflects at least three components:
We can distinguish between these 3 factors,
using multiple recors. In the d13C
record of benthic (bottom) and planktonic (surface water)
foraminifera we would expect a reservoir-change to be
reflected in both planktonics and benthics, and thus no changes in
the surface to deep gradient should occur IF there are no
concurrent changes in productivity. Changes in oceanic productivity
should result in a change in deep-to-surface gradient. Changes in
ocean circulation can be traced by comparing the sea-floor records
from different sites in the oceans.